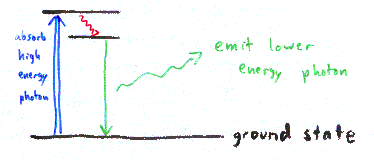
To start with, you need to understand fluorescence:
|
| If you shine light on some molecules, you may see light of a different color emitted from those molecules. This is known as fluorescence. The molecules absorb high-energy light (blue, for example). This increases the energy of the molecules, represented as the top black line in the diagram (an "excited" molecule). Some of the energy from the blue photon is lost internally (represented by the red squiggly arrow in the picture). The molecule then emits a photon with less energy, green in this example. Fluorescein is a common dye that acts in exactly this way, emitting green light when hit with blue excitation light. The color of light emitted is material dependent, and likewise the excitation light wavelength depends on the material. (There are other forms of inelastic scattering; fluorescence is particularly strong.) |
The advantage of fluorescence for microscopy is that you can often attach fluorescent dye molecules to specific parts of your sample, so that only those parts are the ones seen in the microscope. You can also use more than one type of dye. By changing the excitation light, you can cause one type of dye to fluoresce, and then another, to distinguish two different parts of your sample.
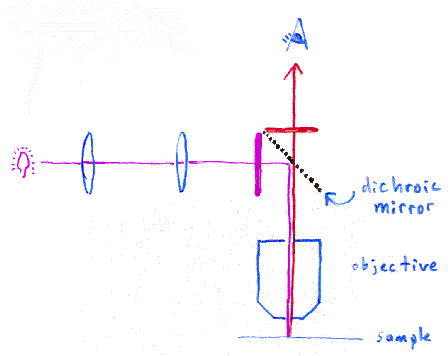
In the picture above, we are supposing the excitation light needs to
be violet, and the emitted light is red. The microscope uses
a special dichroic mirror (or more properly, a "dichromatic
mirror", although this term only seems to be used by purists). This
mirror reflects light shorter than a certain wavelength, and passes
light longer than that wavelength. Thus your eye only sees the emitted
red light
from the fluorescent dye, rather than seeing scattered purple light.
The purple and red bars next to the dichroic mirror represent
additional filters to help prevent the different wavelengths of
light from going the wrong directions.
This particular style of fluorescence microscopy is known as epi-fluorescence, and uses the microscope objective to illuminate the sample (rather than illuminating the sample from the other side, which would be trans-fluorescence).
Well, we're not there just yet, I have to explain one more idea.
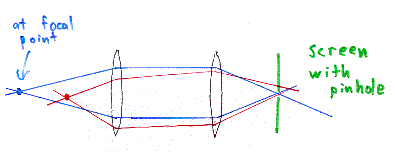
Imagine we have some lenses inside the microscope, that focus light
from the focal point of one lens to another point. This is
represented by the blue rays of light in the above picture.
The red rays of light represent light from another point in the
sample, which is not at the focal point of the lens, but which nonetheless
get imaged by the lenses of the microscope. (Note that the red and
blue rays in the picture are meant to distinguish the two sets of rays,
but they aren't meant to be different wavelengths of light.)
The image of
the red point is not at the same location as the image of the blue point.
(You may remember this from introductory optics, perhaps you have seen a
formula such as 1/s + 1/s' = 1/f for locating the image formed by
a lens. Points don't need to be at the focal point of the lens in order
for the lens to form an image.)
So, we want to just look at the blue point, that is, the point directly at the focus of the lens. If we put a screen with a pinhole at the other side of the lens system, at the image of the blue point, then all of the light from the original blue point will pass through this pinhole. However, most of the light from the red point is still out of focus at this screen, and gets blocked by the pinhole.
This solves one of the problems of regular fluorescence microscopy. Normally, the sample is completely illuminated by the excitation light, so all of the sample is fluorescing at the same time. Of course, the highest intensity of the excitation light is at the focal point of the lens, but nonetheless, the other parts of the sample do get some of this light and they do fluoresce. This contributes to a background haze in the resulting image. Adding a pinhole/screen combination solves this problem. Because the focal point of the objective lens of the microscope forms an image where the pinhole is, these two points are known as "conjugate points" (or alternatively, the sample plane and the pinhole/screen are conjugate planes). The pinhole is conjugate to the focal point of the lens, thus it is a confocal pinhole.
We put all these ingredients together:
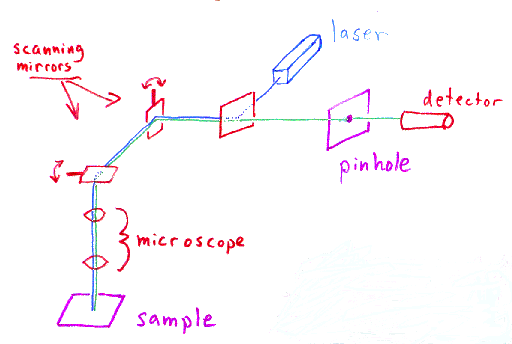
A laser is used to provide the excitation light (in order to get
very high intensities). The laser light (blue) reflects off a dichroic mirror.
From there, the laser hits two mirrors which are mounted on motors;
these mirrors scan the laser across the sample. Dye in the sample
fluoresces, and the emitted light (green) gets descanned by the same mirrors
that are used to scan the excitation light (blue) from the laser.
The emitted light passes through the dichroic and is focused onto the
pinhole. The light that passes through the pinhole is
measured by a detector, ie., a photomultiplier tube.
So, there never is a complete image of the sample -- at any given instant, only one point of the sample is observed. The detector is attached to a computer which builds up the image, one pixel at a time. In practice, this can be done perhaps 3 times a second, for a 512x512 pixel image. The limitation is in the scanning mirrors. Our confocal microscope (from Noran) uses a special Acoustic Optical Deflector in place of one of the mirrors, in order to speed up the scanning. This uses a high-frequency sound wave in a special crystal to create a diffraction grating, which deflects the laser light (actually, the first diffraction peak is used, with the zeroth-order peak being thrown away). By varying the frequency of the sound wave, the AOD changes the angle of the diffracted light, helping scan the sample quickly, allowing us to take 512x480 pixel images 30 times per second. If you want to look at a smaller field of view, our confocal microscope can go even faster (up to 480 frames per second, although I personally find that 240 frames per second is a good practical limit).
By having a confocal pinhole, the microscope is really efficient at rejecting out of focus fluorescent light. The practical effect of this is that your image comes from a thin section of your sample (you have a small depth of field). By scanning many thin sections through your sample, you can build up a very clean three-dimensional image of the sample. Some examples of this are given at this page, looking at emulsions.
Also, a similar effect happens with points of light in the focal plane, but not at the focal point -- emitted light from these areas is blocked by the pinhole screen. So a confocal microscope has slightly better resolution horizontally, as well as vertically. In practice, the best horizontal resolution of a confocal microscope is about 0.2 microns, and the best vertical resolution is about 0.5 microns. I wrote a brief discussion here of the difference between resolution and magnification.
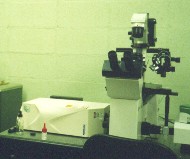 |
The left picture shows our inverted microscope, with a big white
box to the left of it: the white box is the confocal microscope
attachment. As far as the inverted microscope is concerned,
the confocal attachment is just some sort of fancy camera. As
far as the confocal is concerned, the inverted microscope is just
some sort of fancy lens.
The right picture shows the confocal microscope attachment mounted on top of the upright microscope. The confocal microscope attachment shown in these pictures contains the optics for scanning the laser beam, and the pinhole. The confocal also includes a very large box containing electronics, which is not shown in the photographs; there is also a laser, and an SGI computer. |
 |
That's about it. The confocal microscope was invented by Marvin Minsky, who has written a nice summary about this on the web. His original idea was to move the sample in order to scan it, rather than to use scanning mirrors. With the invention of the laser, confocal microscopes became practical.
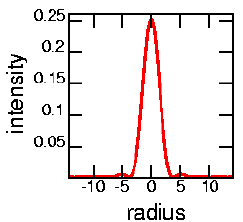
| By the way, you may recall that the image of a point source of light isn't actually a point; due to diffraction, it's actually an Airy disk. The graph to the left shows a plot of the intensity of light as a function of radius; the image is circularly symmetric, as shown at the right. In an ideal world the image of a point would just be a single intense point right at radius=0. The size of the confocal pinhole needs to be matched to the size of the Airy disk. Any smaller, and you are throwing out useful light. Any larger, and you see more out of focus light. For the graph at the left, the pinhole would have a radius of about "4". The image below shows an overexposed picture of an Airy disk, where you can see the secondary ring. |  |

Two articles we wrote that expand on this webpage: