
As part of our research into the nanoscale properties of polymer materials, we develop experimental methods that give us access to different types of material properties such as the glass transition temperature Tg, physical aging, modulus, density, and phase separation. In some cases we are adapting existing methods to give us improved sensitivity at nanoscale dimensions, in other cases we are developing new methods to provide information about how localized polymer properties are perturbed near interfaces. Overall, we are interested in understanding how different material properties that are related to each other in bulk systems are affected and related to each other in nanoscale materials.
Temperature Dependent Perylene Fluorescence as a Probe of Local Polymer Glass Transition Dynamics
Yixuan Han and Connie B. Roth,
Soft Matter 2022, 18, 6094-6104.
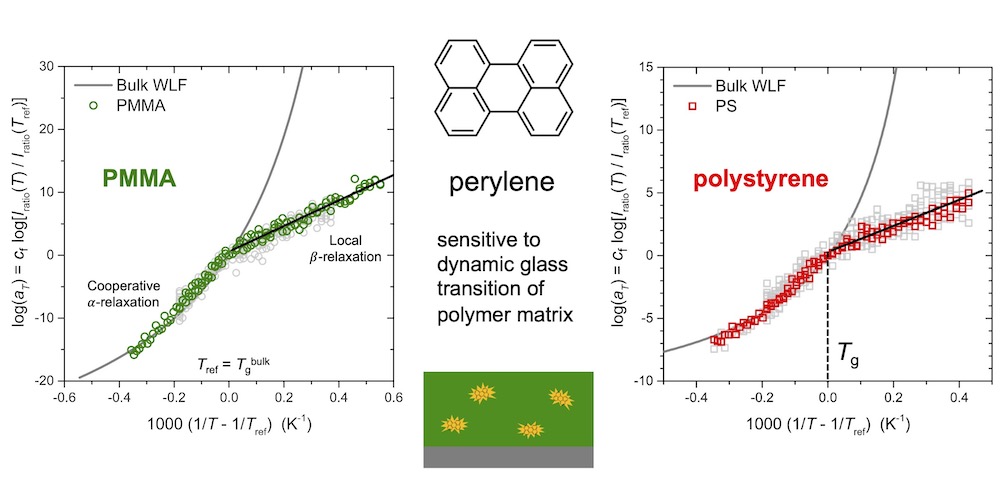
We’ve investigated the temperature dependence of perylene’s fluorescence emission spectrum doped in bulk polymer matrices of poly(methyl methacrylate) (PMMA), polystyrene (PS), poly(2-vinyl pyridine) (P2VP), and polycarbonate (PC) films. Indentifying a temperature-dependent self referencing region (SRR) in the spectra, we show how an intensity ratio can be defined to provide a local measure of the dynamic glass transition temperature Tg. The temperature dependence of the intensity ratio reflects intermolecular collisions of the perylene dye with the surrounding polymers segments. We find it follows a non-Arrhenius temperature depenedence above the glass transition temperature Tg, well described by the bulk Williams–Landel–Ferry (WLF) equation, while transitioning to an Arrhenius temperature dependence below Tg.
In collaboration with Simon Blakey’s group in Chemistry at Emory and grad student Mark Maust, our current efforts are focused on developing this perlyene dye into a new localized measure of the glass transition reflecting a dynamic Tg.
Physically Intuitive Continuum Mechanics Model for QCM: Viscoelasticity of Rubbery Polymers at MHz Frequencies
Yannic J. Gagnon, Justin C. Burton, and Connie B. Roth,
Journal of Polymer Science 2022, 60, 244-257.
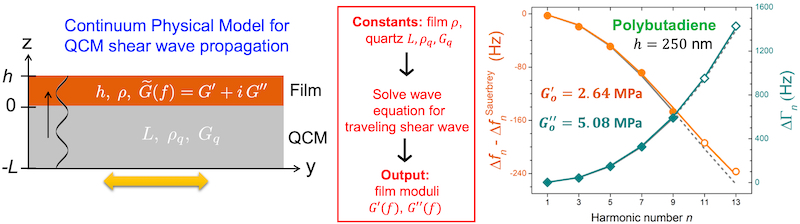
In our first collaboration between the Burton Lab and the Roth Lab, we have developed a continuum physics model that describes the propagation of shear waves through a polymer film caused by the resonance oscillation of a quartz crystal microbalance (QCM) sensor. By solving the resulting set of coupled equations numerically, we can fit the experimental data for the resonance frequency Δfn and dissipation ΔΓn shifts as a function of harmonic number n, over an extended harmonic range approaching film resonance, with zero approximations. This allows us to determine the frequency-dependent modulus of polymer films at MHz frequencies, modeled as linear on a log–log scale. This first paper demonstrates application of this continuum physics model on rubbery polymer films of polybutadiene (PB) and polydimethylsiloxane (PDMS) films, showing excellent agreement with time–temperature shifted rheometry data from the literature. We are now working on applying this method to glassy-rubbery bilayer films.
See: Understanding coupled glass transition dynamics across polymer interfaces
Changes in the Temperature-Dependent Specific Volume of Supported Polystyrene Films with Film Thickness
Xinru Huang and Connie B. Roth,
Journal of Chemical Physics 2016, 144, 234903.
Ellipsometry measures the change in polarization of light as it reflects off of a sample. By modeling the sample with an optical layer model using Fresnel equations, the film thickness h and refractive index n of the film can be determined to great precision. Here we use ellipsometry to measure the temperature-dependent index of refraction of polystyrene (PS) films supported on silicon and investigate the validity of the commonly used Lorentz-Lorenz equation for inferring changes in density or specific volume from very thin films. We conclude that the derivation of the Lorentz-Lorenz equation becomes invalid for very thin films as the film thickness approaches ∼20 nm, and that reports of large density changes greater than ±1% of bulk for films thinner than this likely suffer from breakdown in the validity of this equation or in the difficulties associated with accurately measuring the index of refraction of such thin films. We do observe small shifts of 0.4 ± 0.2%, outside of our experimental error, in the density (inverse of specific volume) for thin films that occur simultaneously in both the liquid and glassy regimes uniformly together. We have since gone on to investigate these effects in a number of different polymers.
See: Nanoscale property changes in thin polymer films
Effect of Adjacent Rubbery Layers on the Physical Aging of Glassy Polymers
Phillip M. Rauscher, Justin E. Pye, Roman R. Baglay, and Connie B. Roth,
Macromolecules 2013, 46, 9806-9817.
We have also developed ellipsometry methods to measure the physical aging rate of polymer films (see below). In this work, we developed fitting methods that would allow us to extract the physical aging rate of thin glassy layers within rubbery-glassy bilayers. In addition, we refined an existing fluorescence method developed by the Torkelson Lab to measure the local glass transition temperature Tg using pyrene dyes covalently bonded to polymer chains. This has allowed us to map profiles in the local glass transition temperature Tg(z) within a number of different systems.
See: Understanding coupled glass transition dynamics across polymer interfaces
and Impact of surface bound chains
Characterization of Phase Separation of Polystyrene / Poly(vinyl methyl ether) Blends Using Fluorescence
Annika Kriisa, Sung S. Park, and Connie B. Roth,
Journal of Polymer Science, Part B: Polymer Physics 2012, 50, 250-256.
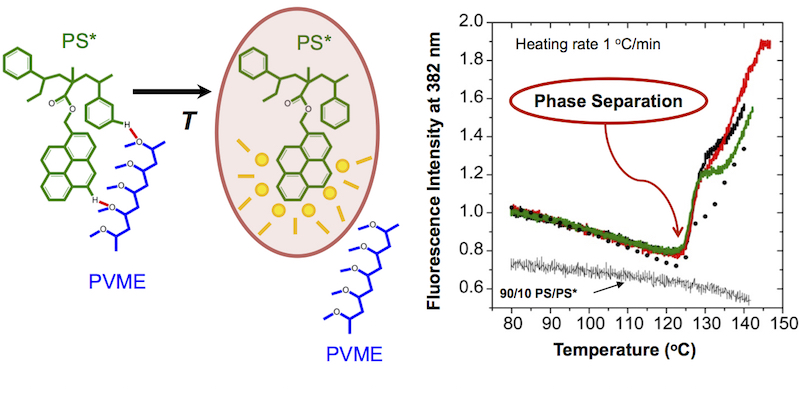
We have characterized for the first time changes in the fluorescence emission spectra of pyrene and anthracene dyes covalently bonded to polystyrene (PS) upon phase separation from poly(vinyl methyl ether) (PVME). The specific chemical structure of the fluorescent labels is found to affect the measured phase separation temperature T_s, with fluorophores covalently attached in closer proximity to the PS backbone identifying phase separation a few degrees earlier. The sharp increase in fluorescence intensity upon phase separation that occurs for all fluorophores with little change in spectral shape is consistent with a mechanism of static fluorescence quenching resulting from the specific interaction with a nearby quenching molecular unit. Based on recent work that has identified a weak hydrogen bond occurring between the aromatic hydrogens of PS and the ether oxygen of PVME, we believe a similar weak hydrogen bond is likely occurring between the PVME oxygen and the aromatic dyes providing a local (few nanometer) sensitivity to phase separation. Our observations indicate that fluorescence is sensitive to the early initiation of phase separation. In comparing similar dyes, fluorophores in which the dye is covalently bonded closer to the PS backbone denote phase separation on average 3 °C earlier than those fluorophores in which the dye is located further away on a covalent tether. As the PVME component moves further away from the PS component upon phase separation, local breaking of these weak hydrogen bonds connecting the PVME oxygen with the aromatic rings of PS and the aromatic dyes, leads to the observed increase in fluorescence intensity. This reliable and reproducible method of measuring the phase separation temperature T_s allowed us to quantify how the presence of electric fields shifted the miscibility of PS/PVME blends.
See: Tuning miscibility of polymer blends with electric fields
Streamlined Ellipsometry Procedure for Characterizing Physical Aging Rates of Thin Polymer Films
Elizabeth A. Baker, Perla Rittigstein, John M. Torkelson, and Connie B. Roth,
Journal of Polymer Science, Part B: Polymer Physics 2009, 47, 2509-2519.
We have developed a new method of characterizing the physical aging rate of thin polymer films in an efficient manner using ellipsometry. Ellipsometry measures the change in polarization of light as it is reflected from the sample. By using Fresnel’s equations, it is possible to determine the film thickness and index of refraction of a very thin layer supported on a known substrate (typically silicon). Using visible light from 400-1000 nm, we are able to measure the film thickness to within a fraction of a nanometer. In comparison to other methods available for characterizing the structural relaxation of thin films, this method has the advantage that it can be easily applied to different polymers with varying chemical structure. The structural relaxation of the sample is characterized by measuring the decrease in film thickness and increase in index of refraction resulting from the slow increase in density of the material as a function of time. We quantify the physical aging rate β from the time-dependent decrease in the film thickness as a function of logarithmic time: β = − d(h/h0) / d(log t). This streamlined ellipsometry method for characterizing physical aging in polymer films has enabled a number of studies investigating changes in polymer glass stability in our lab and others.
See: Stress during vitrification impacts stability of polymer glasses
and Nanoscale property changes in thin polymer films